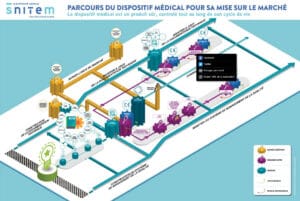
Tous les dispositifs médicaux doivent obligatoirement être certifiés selon une réglementation stricte. En Europe, la conformité à cette réglementation est matérialisée par la délivrance du marquage CE médical qui garantit que le dispositif médical répond aux exigences de sécurité et de bénéfice clinique fixées. Les points essentiels de la réglementation des dispositifs médicaux.
Les étapes clés
Différentes étapes jalonnent le parcours d’un dispositif médical, de sa conception à sa mise sur le marché, en passant par les études, évaluations, audits etc.
Chaque étape doit répondre à des exigences réglementaires strictement encadrées.
LES 4 ÉTAPES DU MARQUAGE CE MÉDICAL
Le marquage CE médical traduit la conformité d’un dispositif médical aux exigences essentielles de sécurité et de bénéfice clinique fixées par la règlementation européenne. Il constitue un préalable nécessaire à la mise sur le marché d’un DM au sein de l’Union européenne.
Les exigences générales en matière de sécurité et de performance sont identiques pour tous les produits, mais le mode de démonstration de la conformité à ces exigences sera d’autant plus contraignant que la classe de risque du DM est élevée.

- Le fabricant choisit librement un organisme notifié (ON) parmi les ON européens habilités pour ses catégories de DM.
- S’il s’agit d’un DM de classe I, il auto-certifie son produit.

L’ÉVALUATION DU TYPE
- Vérification que le produit répond aux exigences de sécurité et de performance.
- Portant sur la conception, la conformité aux normes et référentiels pré-clinique et clinique, le processus de production.
- Compilation des éléments dans la documentation technique.
L’ÉVALUATION DU SYSTÈME DE MANAGEMENT DE LA QUALITÉ (SMQ)
- Évaluation de la capacité de l’entreprise à reproduire le type en séries en assurant la conformité aux exigences essentielles.
- Mise en place par le fabricant d’un SMQ décrivant tous les process associés (gestion des modifications, gestion des risques, procédés de fabrication…) et de conformité.

- Certificat de marquage CE médical délivré par l’organisme notifié pour une durée de 5 ans maximum.

Une fois sur le marché européen, les fabricants de dispositifs marqués CE continuent d’être évalués par l’organisme notifié :
- Audit annuel de surveillance ;
- Audit approfondi lors de modifications ou du renouvellement du marquage CE ;
- Audits inopinés (au moins tous les 5 ans) ;
- Évaluation approfondie de la documentation technique lors de modifications ou du renouvellement
du marquage CE.
En France, la qualité et la sécurité (matériovigilance, contrôle, inspection) de tous les DM mis sur le marché sont également surveillées par l’Agence nationale de sécurité du médicament et des produits de santé (ANSM).
À SAVOIR
LES DM SONT CLASSÉS SELON LEUR RISQUE :
- Classe I : classe de risque la plus faible (compresses, thermomètres, lunettes, lits médicaux…).
- Classe IIa : risque potentiel modéré/mesuré (lentilles de contact, produits d’obturation dentaires…).
- Classe IIb : risque potentiel élevé/important (pompes à perfusion, systèmes de radiothérapie, hémodialyseurs, préservatifs, produits de désinfection des lentilles…).
- Classe III : classe de risque la plus élevée (implants mammaires, stents, prothèses de hanche, stérilets, stimulateurs cardiaques…).
POUR EN SAVOIR PLUS
Consulter le parcours du dispositif médical pour sa mise sur le marché ; ce parcours plus complet mais aussi plus complexe montre le rôle du fabricant et des autorités compétentes tout au long du cycle de vie du dispositif médical.
COMPRENDRE LES ÉVOLUTIONS DU RÈGLEMENT EUROPÉEN EN UN COUP D’OEIL
La réglementation qui encadre le secteur du dispositif médical (DM) évolue sans cesse. Dernier changement en date : la mise en application en mai 2021 du nouveau règlement européen 2017/745 dont le champ d’application est élargi et qui réglemente toutes les étapes clés de la mise sur le marché des DM. Voici les principales évolutions induites.

> Précision de la définition du dispositif médical et augmentation du champ d’application, par exemple :
- Exclusion des produits contenant des substances ou organismes biologiques ;
- Inclusion des produits à visée non médicale…
> Évolution des règles de classification des dispositifs :
- Nombreuses reclassifications à la hausse par exemple pour les logiciels, certains implants, les dispositifs en circuit fermé…
> Nouvelles exigences applicables (par exemple, en matière de cyber sécurité), nouvelles procédures (avec, par exemple, le recours à des expertises spécifiques avec des panels d’experts européens…) et référentiels techniques (spécifications communes).
> Révision complète des documentations techniques pour tous les produits.

> Évaluation clinique renforcée avant et après commercialisation.
> Augmentation du niveau de démonstration.
> Restriction forte pour l’utilisation des données d’équivalence avec un autre DM.
> Investigations cliniques obligatoires pour les DM implantables.

> Cahier des charges des ON renforcé (par exemple en matière de compétences, via des visites inopinées…).
> ON placés sous contrôle européen (harmonisation des pratiques).

> Vigilance et surveillance renforcées :
- Base de données européennes des incidents ;
- Délais de notification raccourcis ;
- Process optimisés ;
- PSUR (rapport périodique actualisé de sécurité).
> Obligations des différents opérateurs économiques précisées :
- Fabricant ;
- Distributeur ;
- Mandataire ;
- Importateur ;
- Personne visée à l’article 22 ;
- Personne chargée de veiller au respect de la réglementation.

> Mise en place de l’identifiant unique pour chaque dispositif.
> Mise en oeuvre d’Eudamed, la base de données européenne accessible au public (informations sur les produits, résumé de caractéristiques pour les DM classe III, résumé des rapports d’investigation clinique, rapport de tendances, rapport de synthèse, informations de sécurité etc.).

> Règlement d’application directe ne nécessitant pas de transposition dans les droits nationaux.
> Groupe de coordination des DM : publication de nombreux guides d’interprétation.